
Generando los conjuntos de datos

En posts anteriores mostré como el análisis estadístico de las secuencias del SARS-Cov2 puede ser usado para encontrar diversas adaptaciones dentro del genoma, así como la temporalidad del mismo. Una parte fundamental del análisis es la generación de un conjunto de datos que consiste en la frecuencia de fragmentos de la secuencia. Para la generación de este conjunto de datos se cuentan con dos fuentes primarias. El repositorio del GISAID que nació con el objetivo de compartir secuencias genómicas de influenza y el repositorio de recursos del SARS-Cov2 del NCBI. Para acceder a los datos genómicos del GISAID es necesario estar afiliado a una institución de educación superior o dedicada a la investigación. Por lo que acceder a este repositorio es más complicado si no se poseen esos requerimientos. Mientras que en el caso del NCBI la descarga de datos no requiere algún tipo de registro, por lo que es la base de datos que he usado.
Usualmente, las secuencias biológicas se encuentran en formato FASTA, que es un formato de texto que guarda algunas características de la secuencia como un identificador único, el organismo, origen, país de origen, además de la secuencia. Para las secuencias de ADN/RNA la secuencia se encuentra constituida por solo cuatro caracteres distintos que representan cada uno de los nucleótidos del ADN/RNA. Cuando el genoma no puede ser reconstruido de forma precisa, se pueden encontrar otros caracteres que indican el probable tipo de base en esa localización o que en esa posición hay una base de algún tipo.
Para la generación de la base de datos solo dos condiciones fueron impuestas, que fuera el genoma completo y que el genoma solo se encontrara compuesto de 4 caracteres únicos. Es decir, se seleccionaron los genomas completos con la mejor confianza en su secuenciación. Eso resulto en alrededor de 1.3 millones de genomas únicos con lo reportado hasta septiembre del 2022.
Dependiendo de los recursos computaciones disponibles, hay algunas formas en las que se deben de pretratar las secuencias antes de generar el conjunto de datos final. Si es posible cargar el archivo completo en memoria RAM, entonces no hay ningún tipo de pretratamiento. De lo contrario se divide el archivo en fragmentos de tamaño más pequeño que permitan su manejo. Cada archivo termina conteniendo un fragmento de todos los genomas descargados.
def BatchIterator(Iterator, BatchSize):
"""Returns lists of length batch_size.
This can be used on any iterator, for example to batch up
SeqRecord objects from Bio.SeqIO.parse(...), or to batch
Alignment objects from Bio.AlignIO.parse(...), or simply
lines from a file handle.
This is a generator function, and it returns lists of the
entries from the supplied iterator. Each list will have
batch_size entries, although the final list may be shorter.
"""
entry = True # Make sure we loop once
while entry:
batch = []
while len(batch) < BatchSize:
try:
entry = next(Iterator)
except StopIteration:
entry = None
if entry is None:
# End of file
break
batch.append(entry)
if batch:
yield batch
Sequences = SeqIO.parse(open(seqDataDir), "fasta")
total = 0
for i, batch in enumerate(BatchIterator(Sequences, 35000)):
filename = sequencesFrags + "group_%i.fasta" % (i + 1)
sequenceIndex = GetFilteredSeqsIndex(batch)
newbatch = [batch[k] for k in sequenceIndex]
total = total + len(newbatch)
with open(filename, "w") as handle:
count = SeqIO.write(newbatch, handle, "fasta")
print("Wrote %i records to %s" % (count, filename))
print(total)Posteriormente, cada secuencia debe de ser subdividida en fragmentos de longitud k y se calcula la frecuencia de cada uno de los fragmentos. Para acelerar el proceso se paraleliza el cálculo de las frecuencias y se añaden las frecuencias de los diferentes tamaños de fragmento de forma horizontal. Una vez terminado este fragmento de la base de datos de genomas, se añaden las demás frecuencias de forma vertical al fragmento antes analizado. Esto resulta en una base de datos de 1.3 mill x 320.
def SplitString(String,ChunkSize):
'''
Split a string ChunkSize fragments using a sliding windiow
Parameters
----------
String : string
String to be splitted.
ChunkSize : int
Size of the fragment taken from the string .
Returns
-------
Splitted : list
Fragments of the string.
'''
try:
localString=str(String.seq)
except AttributeError:
localString=str(String)
if ChunkSize==1:
Splitted=[val for val in localString]
else:
nCharacters=len(String)
Splitted=[localString[k:k+ChunkSize] for k in range(nCharacters-ChunkSize)]
return SplittedEl tamaño de la base de datos resulta del número de secuencias disponibles (1.3millones hasta septiembre del 2022) y la frecuencia de fragmentos únicos. Dado que son 4 bases distintas, el número de combinaciones de 2 elementos es 16, 64 para tres y 256 para 4, sumando resulta en 320 fragmentos únicos. Esta base de datos puede ser guardada como tal y es posible obtener tanto las diferentes oscilaciones como los diferentes conjuntos del modelo de aprendizaje automático.
def CountUniqueElements(UniqueElements,String,Processed=False):
'''
Calculates the frequency of the unique elements in a splited or
processed string. Returns a list with the frequency of the
unique elements.
Parameters
----------
UniqueElements : array,list
Elements to be analized.
String : strting
Sequence data.
Processed : bool, optional
Controls if the sring is already splitted or not. The default is False.
Returns
-------
localCounter : array
Normalized frequency of each unique fragment.
'''
nUnique = len(UniqueElements)
localCounter = [0 for k in range(nUnique)]
if Processed:
ProcessedString = String
else:
ProcessedString = SplitString(String,len(UniqueElements[0]))
nSeq = len(ProcessedString)
UniqueDictionary = UniqueToDictionary(UniqueElements)
for val in ProcessedString:
if val in UniqueElements:
localPosition=UniqueDictionary[val]
localCounter[localPosition]=localCounter[localPosition]+1
localCounter=[val/nSeq for val in localCounter]
return localCounter
def CountUniqueElementsByBlock(Sequences,UniqueElementsBlock,config=False):
'''
Parameters
----------
Sequences : list, array
Data set.
UniqueElementsBlock : list,array
Unique element collection of different fragment size.
config : bool, optional
Controls if the sring is already splitted or not. The default is False.
Returns
-------
Container : array
Contains the frequeny of each unique element.
'''
Container=np.array([[],[]])
for k,block in enumerate(UniqueElementsBlock):
countPool=mp.Pool(MaxCPUCount)
if config:
currentCounts=countPool.starmap(CountUniqueElements, [(block,val.seq,True )for val in Sequences])
else:
currentCounts=countPool.starmap(CountUniqueElements, [(block,val.seq )for val in Sequences])
countPool.close()
if k==0:
Container=np.array(currentCounts)
else:
Container=np.hstack((Container,currentCounts))
return Container
Sin embargo, el tamaño de esta base de datos puede ser muy grande y prevenir su uso, por lo que un esquema adicional de normalización permite guardar los datos ocupando menos espacio. En este caso cada conjunto de fragmentos se normaliza a valores entre 0 y 1. Esta nueva base de datos es capaz de obtener los diferentes conjuntos resultantes del modelo de redes neuronales, pero es incapaz de obtener las oscilaciones del modelo de la serie de tiempo. Por lo que para evaluar la dinámica a largo tiempo, el conjunto de datos normalizados puede no ser una buena opción.
def CountUniqueElementsByBlock(Sequences,UniqueElementsBlock,config=False):
'''
Parameters
----------
Sequences : list, array
Data set.
UniqueElementsBlock : list,array
Unique element collection of different fragment size.
config : bool, optional
Controls if the sring is already splitted or not. The default is False.
Returns
-------
Container : array
Contains the frequeny of each unique element.
'''
Container=np.array([[],[]])
for k,block in enumerate(UniqueElementsBlock):
countPool=mp.Pool(MaxCPUCount)
if config:
currentCounts=countPool.starmap(CountUniqueElements, [(block,val.seq,True )for val in Sequences])
else:
currentCounts=countPool.starmap(CountUniqueElements, [(block,val.seq )for val in Sequences])
countPool.close()
if k==0:
Container=np.array(currentCounts)
else:
Container=np.hstack((Container,currentCounts))
return Container.astype(np.float16)
Estos métodos para la generación de los datos resultan en un conjunto de datos de 9gb aproximadamente, para el conjunto de datos sin normalizar, mientras que el segundo conjunto de datos es de 3gb aproximadamente.
Visualizando la trayectoria del virus.
Una de las constantes dentro del genoma del virus es su constante mutación, estas mutaciones pueden observarse como el cambio en el contenido de los diferentes nucleótidos. Dado que estos cambios se relacionan entre sí, es posible observar la trayectoria de adaptación que sigue el virus. Por ejemplo, al graficar el contenido de cada uno de los nucleótidos obtenemos el siguiente patrón.
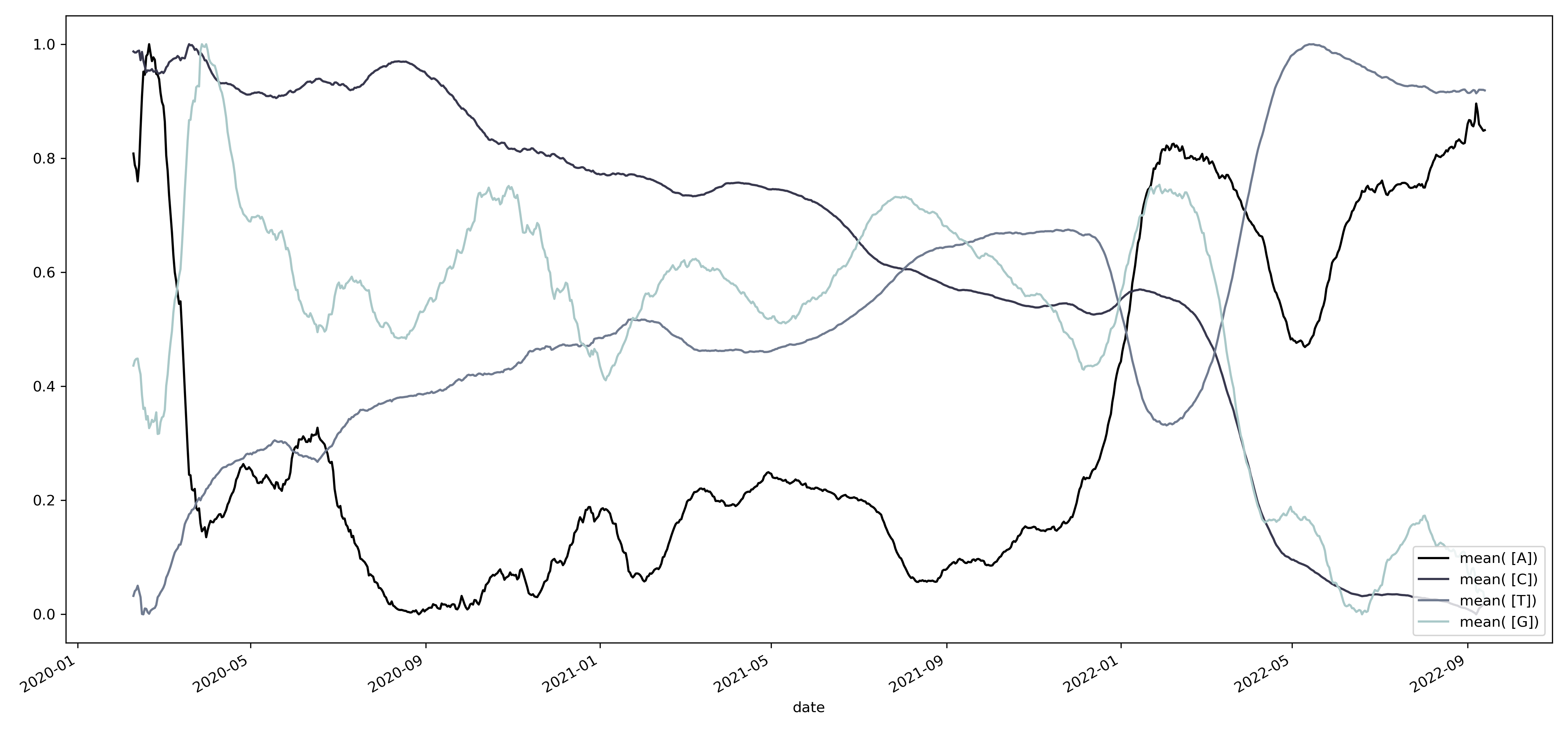
Podemos cambiar la escala temporal al agrupar los datos por año, encontrando una serie de oscilaciones que se repiten a lo largo del tiempo.
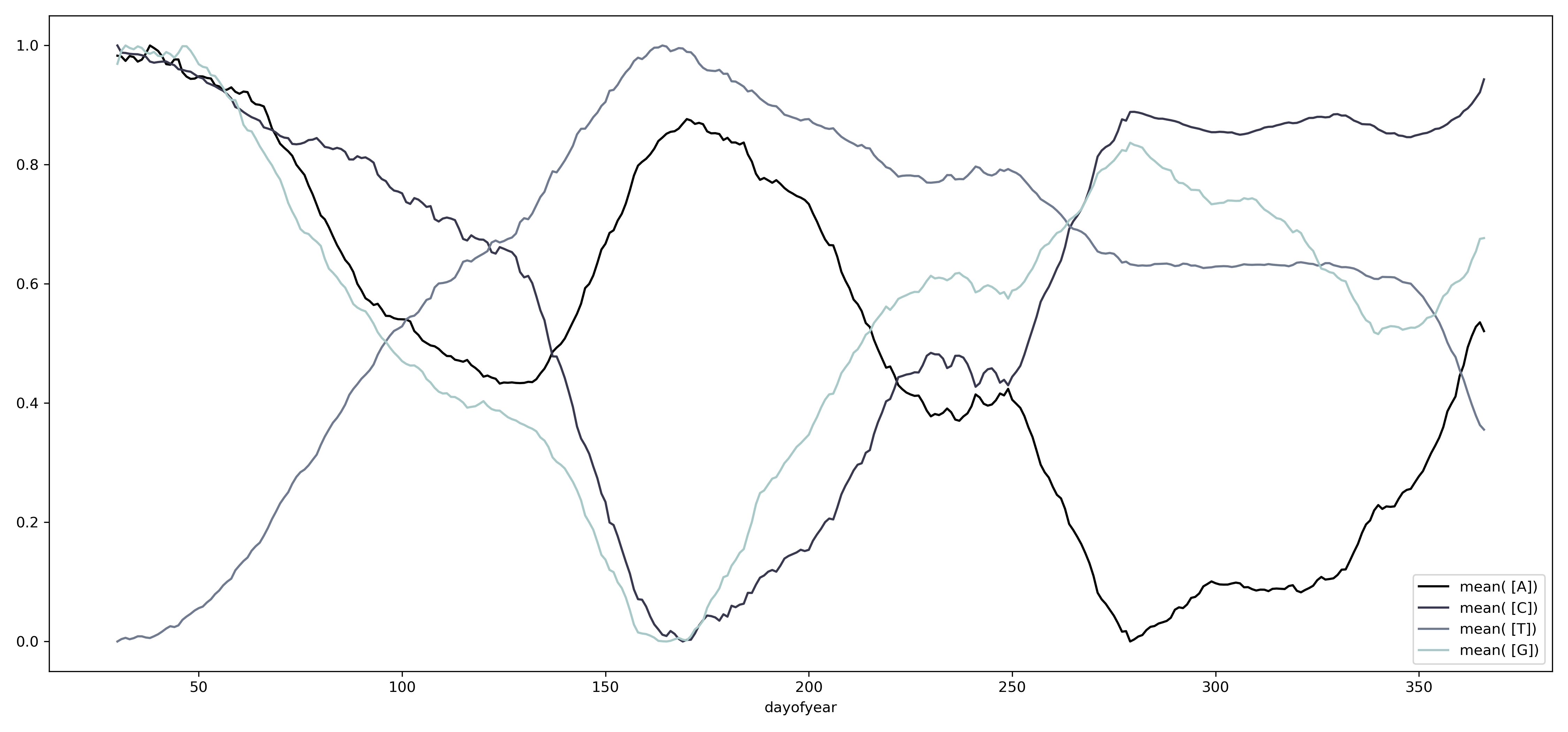
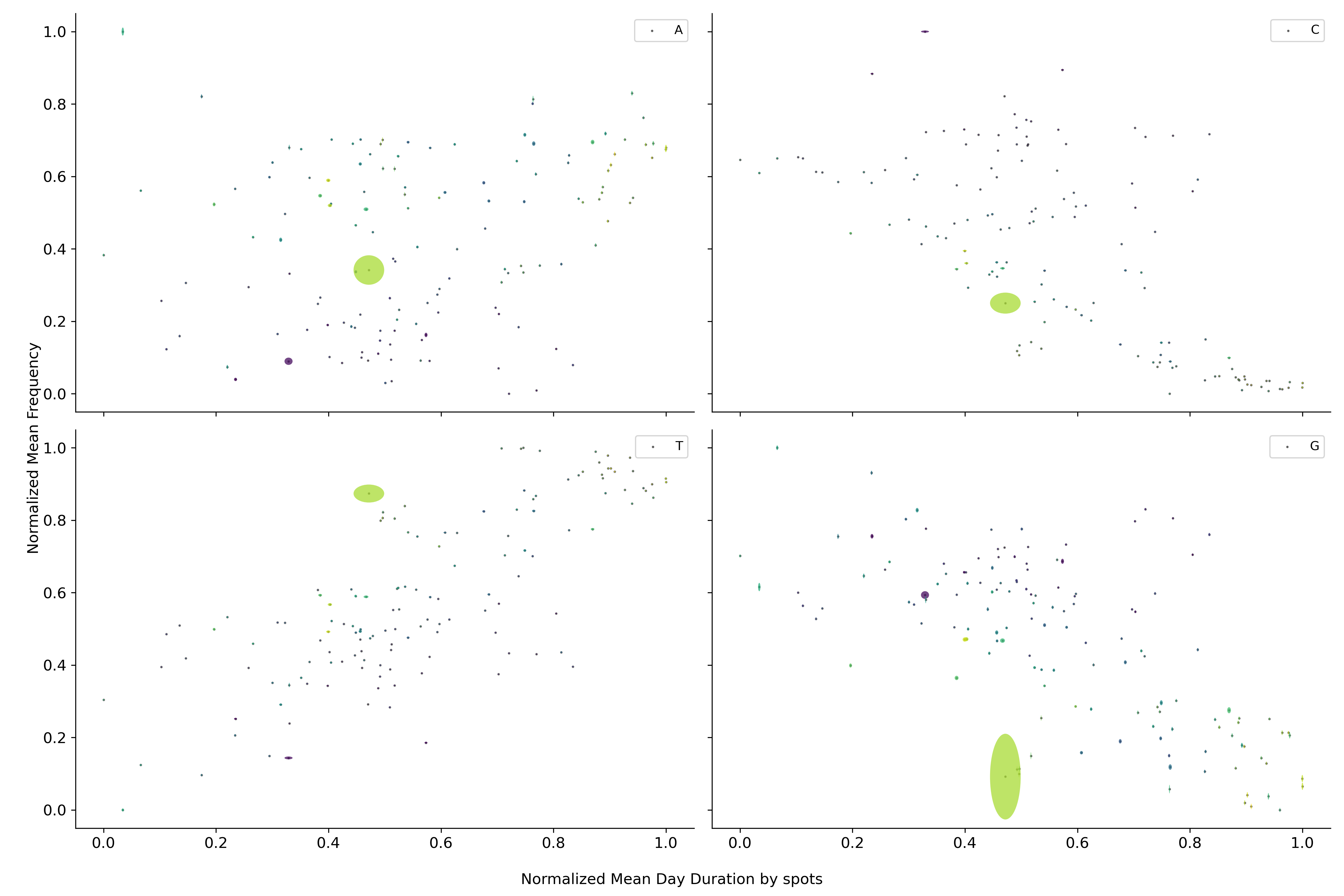
Cambiando la escala temporal a la duración del día, se puede observar que el contenido de los diferentes nucleótidos parece circular alrededor de un punto medio. Este fenómeno también se puede observar para las diferentes combinaciones de nucleótidos.
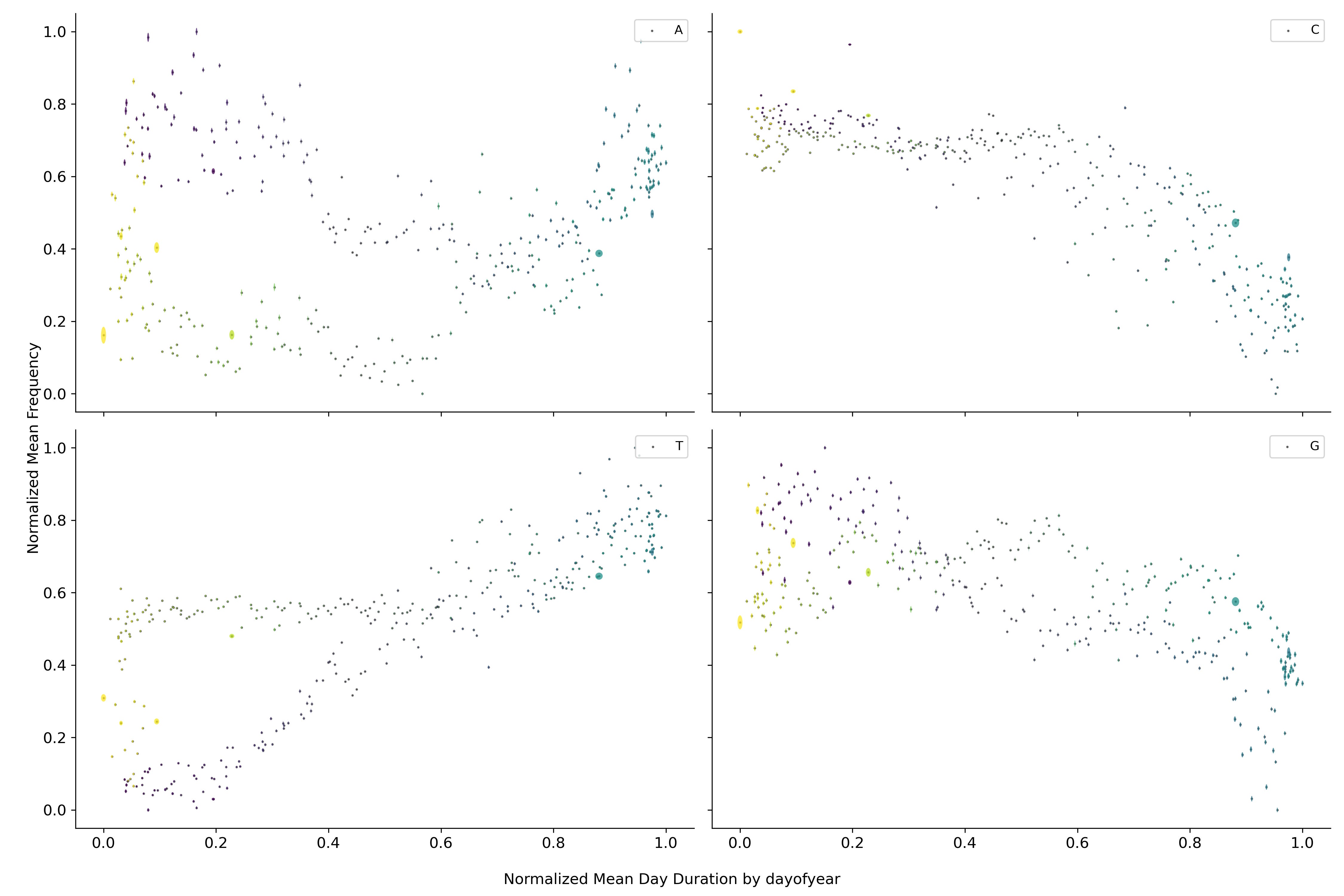
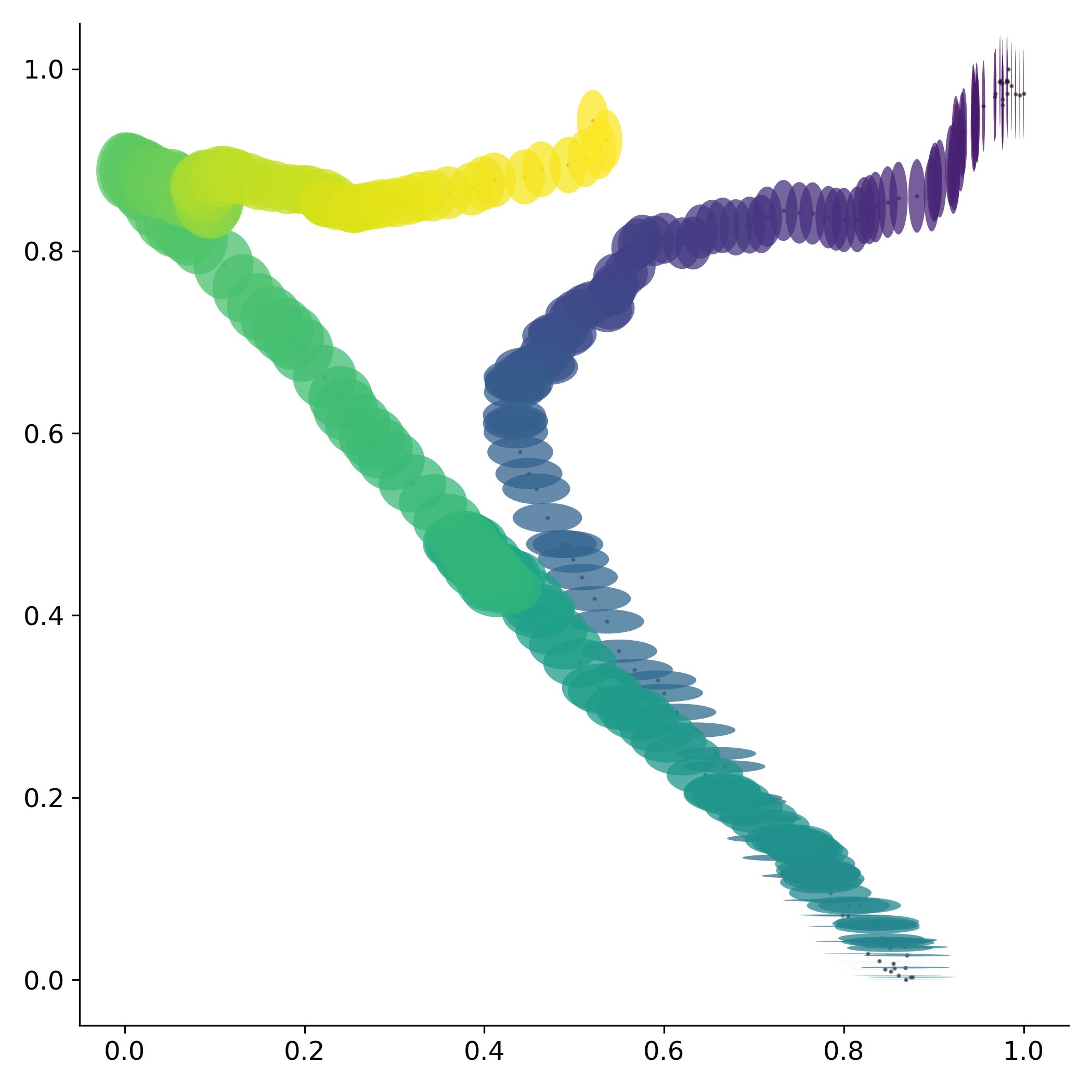
Regresando a la duración de la pandemia se observa que los nucleótidos dejan de orbitar alrededor de un punto medio si no que parecen dirigirse hacia algún punto en particular. Fenómeno que se repite nuevamente para conjuntos de fragmentos más grandes.
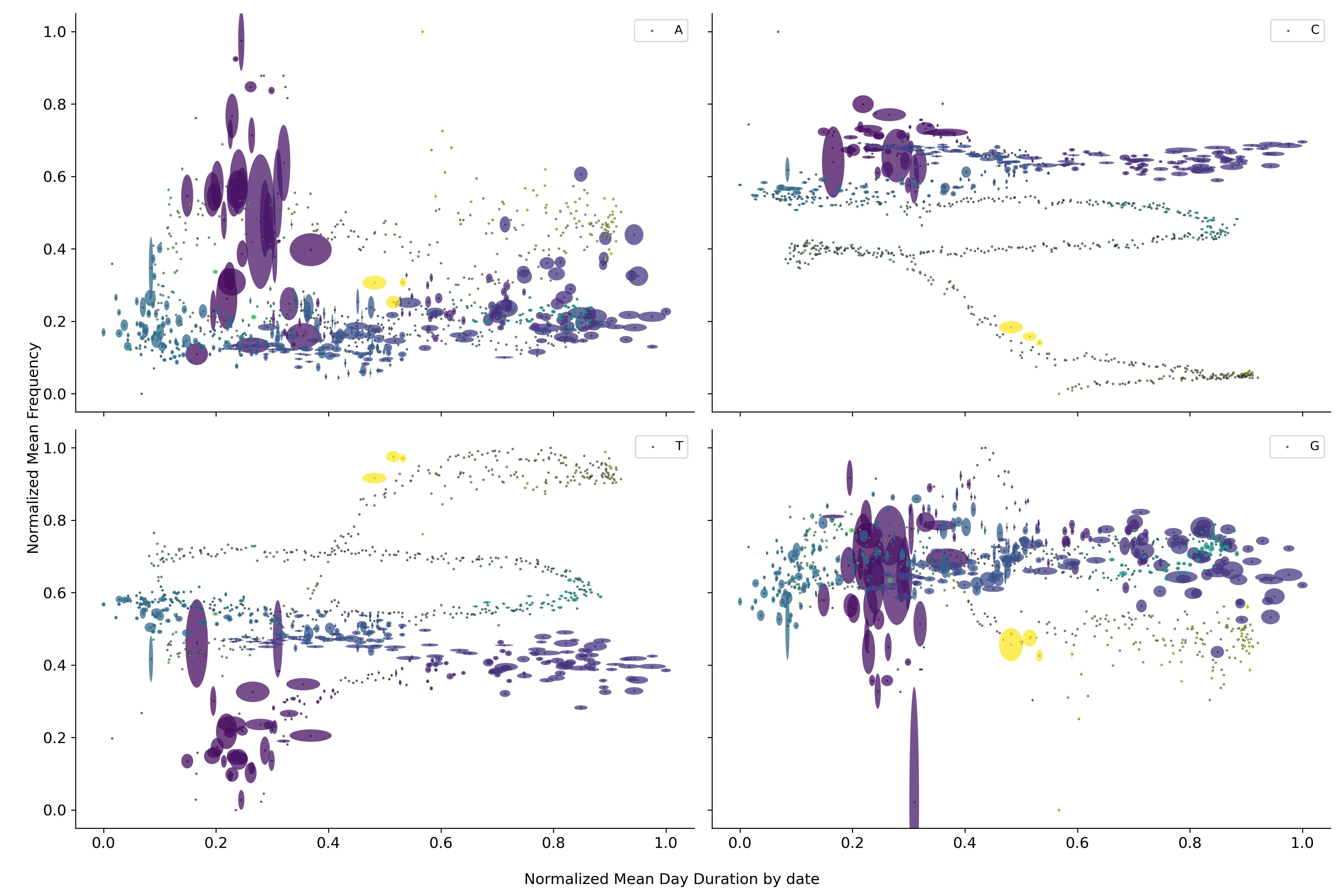
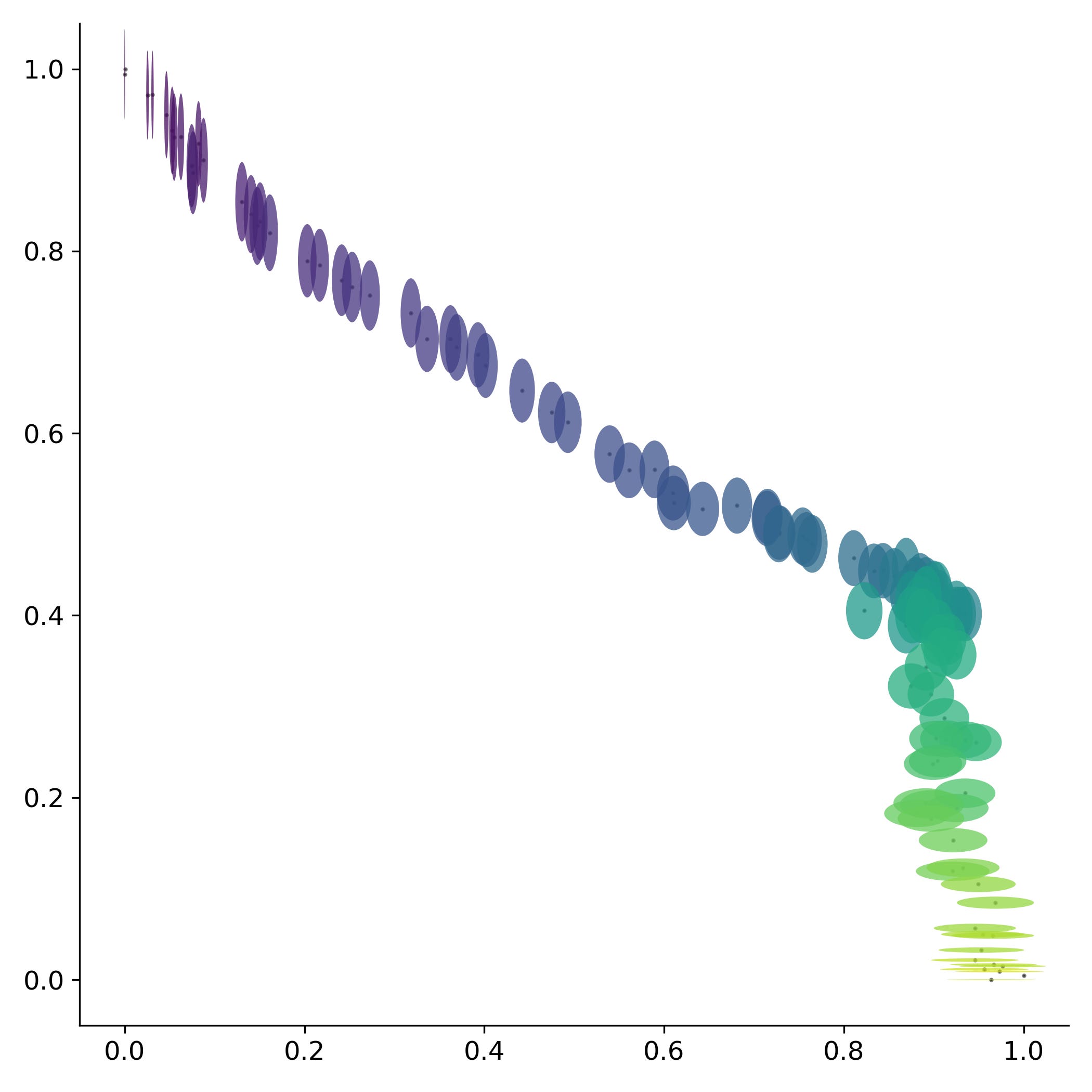
Esta trayectoria sugiere que el virus se está dirigiendo hacia algún punto en específico de convergencia. Sin embargo, el calendario estándar parece no ser una buena medida del tiempo para evaluar los cambios dentro del virus. Por lo que si en su lugar se utilizan el número de manchas solares, las cuales aproximan la ubicación dentro del ciclo solar, se observa una sola trayectoria. Fenómeno que se repite nuevamente para conjuntos de fragmentos más grandes.
Esto nos permite aproximar una serie de escalas temporales dentro de la dinámica del SARS-Cov2. Una dinámica anual gobernada por los cambios en la radiación solar anual en una localización geográfica que puede ser aproximada por la duración del día. Una dinámica a largo plazo gobernada por los cambios en radiación dentro del ciclo de Schwabe que puede ser aproximada por el número de manchas solares. Y probablemente también exista una dinámica de carácter circadiana que esté gobernada por los cambios de radiación solar que ocurren durante un lapso de 24 horas.
Aunque los anteriores conjuntos de datos son capaces de describir la dinámica del SARS-Cov2, el uso de los modelos de aprendizaje profundo fueron los que permitieron encontrar la dinámica codificada dentro de los datos. En el siguiente post mostraré como se puede implementar el modelo, así como diferentes arquitecturas del modelo, su efecto en los resultados y el número mínimo de datos necesarios para obtener los diferentes conjuntos.
El plan
El objetivo de este blog es el de conseguir fondos para mejorar y perfeccionar los métodos de análisis expuestos en “Aplications of sliding sampling to biologiocal sequences”, además de una serie de post en medium y otros análisis diversos. Puedes encontrar un índice del código y modelos propuestos en el siguiente enlace.
El desarrollo de esta metodología puede ofrecer una caracterización rápida de patógenos emergentes y otras enfermedades. Durante el desarrollo del proyecto, todos los recursos, desde código, conjuntos de datos, y ejemplos de como usar, se publicarían de modo periódico y libre.
El apoyar proyectos científicos fuera de la academia es una actividad cada vez más común, algo que se denomina ciencia ciudadana. Sin embargo, el obtener fondos para estos proyectos continúa siendo un factor limitante. El apoyar este tipo de proyectos traería a la luz nuevas ideas o ideas previamente ignoradas.
Evite las grandes aglomeraciones cerca de los puntos estacionarios de radiación solar, medio día y verano o invierno. Mejore el tiempo de exposición a la radiación solar que tiene diariamente. Y nos vemos en el siguiente.