

Reducción de la dimensionalidad y clasificación de secuencias biológicas.
Reducción de la dimensionalidad y clasificación de secuencias biológicas.

En un post anterior presente una forma de analizar las secuencias del SARS-Cov2 usando métodos estadísticos y series de tiempo. Estos métodos utilizan la frecuencia de fragmentos de la secuencia como una representación resumida de la secuencia. Por lo que el orden y el contexto de la secuencia se pierde, sin embargo, es posible observar oscilaciones que están relacionadas con la aparición de nuevas variantes. No obstante, al aumentar el tamaño del fragmento, el número de posibles fragmentos crece complicando el análisis.

Esta dificultad, que nace de refinar el análisis, genera conjuntos de datos con muchas características. Es decir, cada secuencia es descrita por 4, 16, 64 y más componentes únicos. Todas estas características o componentes generan conjuntos de datos de alta dimensionalidad. Computacionalmente, algunos tipos de análisis pueden restringirse debido al tamaño de los datos. Además de dificultar la visualización de los mismos.
Reducción de la dimensionalidad
Una forma sencilla de analizar conjuntos de datos de alta dimensionalidad es mediante el uso de técnicas de reducción de la dimensionalidad como el PCA. El PCA o análisis de componente principal proyecta los datos a un espacio de menor dimensiones, el cual maximiza la variabilidad de los datos. Una analogía de la operación que realiza el PCA sería la sombra de un objeto. El conjunto de datos original es el objeto, mientras que la sombra del objeto seria la proyección por PCA.

Si tomamos la proyección por PCA de la frecuencia de fragmentos de forma anidada, se empieza a observar una segmentación dentro de los datos.
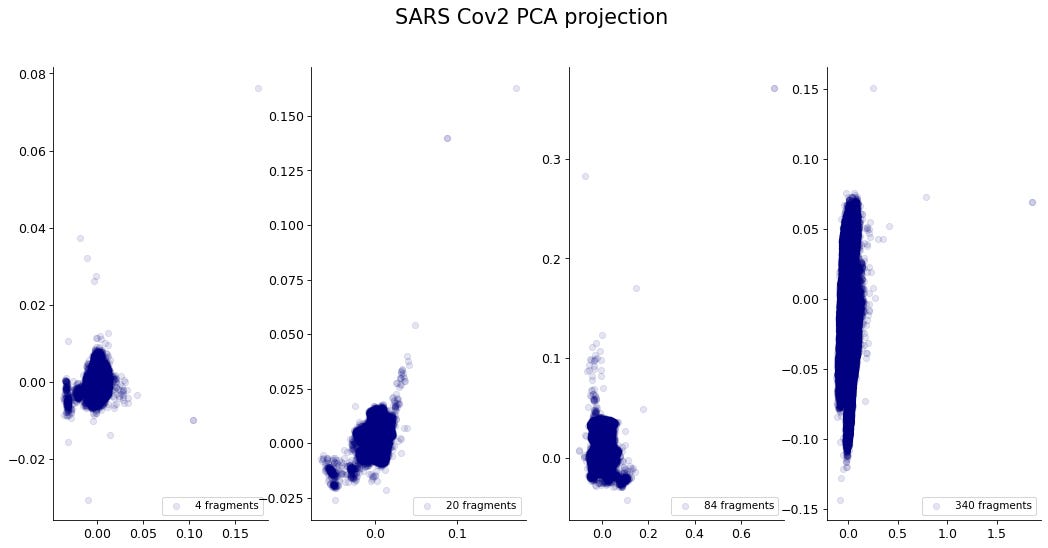
Probablemente este comportamiento se debe a la estructura de árbol de decisiones dentro de los datos. Cada fragmento se encuentra sobrelapado a los demás, por lo que en combinación se restringe la frecuencia de cada fragmento. Este fenómeno se puede observar mejor al cambiar solo un carácter en la secuencia del SARS-Cov2. Este simple cambio se ve amplificado a una gran variedad de fragmentos dentro de la secuencia.

El agrupamiento característico que resulta de la proyección por PCA muestran que hay patrones que se repiten y que pueden ser aislados. Sin embargo, una de las desventajas del PCA es que hay información que se pierde durante la proyección. Generalmente una proyección de baja dimensionalidad puede perder la mayoría de la información dentro del conjunto de datos. Por lo que para prevenir la perdida de información es necesario usar una mejor técnica de reducción de la dimensionalidad.
Redes neuronales y modelos generativos
Una mejor técnica para reducir la dimensionalidad es el uso de redes neuronales. Particularmente el empleo de autocodificadores variacionales o VAEs. Los VAEs son redes neuronales que se componen de dos componentes, un codificador y un decodificador. El codificador realiza la operación de reducir la dimensionalidad de los datos, mientras que el decodificador reconstruye el dato original. Por lo que los VAEs se entrenan utilizando los mismos datos de entrada como de salida.

Una característica importante de la reducción de dimensionalidad obtenida por un VAE es que cada dimensión representa un atributo dentro de los datos. La naturaleza de este atributo depende de la naturaleza de los mismos y la capacidad de la red para codificar este patrón. Aplicando un VAE a los datos anteriores resulta en una estructura de múltiples grupos, los cuales se ordenan de acuerdo a la fecha de aislamiento de las secuencias.

Particularmente este atributo se encuentra codificado en una sola de las dimensiones de la proyección, y este componente temporal parece ajustarse mejor a una escala anual. Por lo que el modelo podría predecir puntos a lo largo del tiempo donde sea posible aislar un mayor número de variantes debido al cambio de un grupo a otro por el virus.
Puntos de alta mutación viral y patrones repetidos
Al ordenarse las secuencias de forma anual se observa que hay periodos donde la secuencia viral pasa de un grupo a otro. Estos puntos de transición podrían mostrar puntos donde el virus adapta su secuencia y, por lo tanto, es posible aislar un mayor número de variantes del virus. Una forma simple de evaluar estos puntos de transición es mediante medir el número de variantes que son identificadas por día del año. Este análisis muestra que en los puntos de transición hay un mayor número de variantes únicas aisladas.
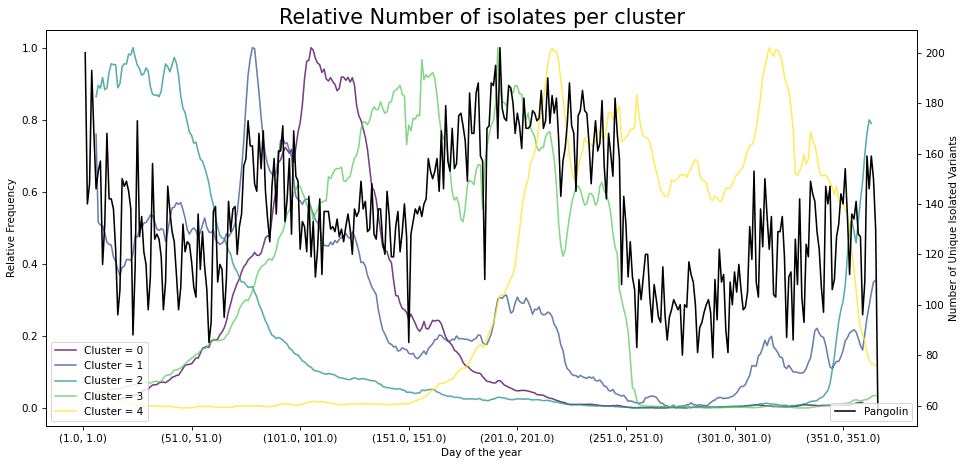
Además, al evaluar la primera y última vez que una variante es aislada se observan puntos de convergencia. Particularmente en regiones donde se pasa de un grupo a otro.

Estos diferentes grupos de secuencias podrían representar una serie de patrones que se repiten dentro de la secuencia del SARS-Cov2. Estos patrones podrían representar la forma en la que el virus se adapta a los cambios en su ambiente. Uno de los cambios identificados anteriormente es la perdida de la amplificación de gen S. Esta característica se ha utilizado para la clasificación de diferentes variantes mediante PCR. Si hay cambios recurrentes dentro de la secuencia, esta característica debería de aparecer y desaparecer a lo largo del tiempo. Al evaluar la localización de variantes positivas y negativas al gen S se puede observar que estas se encuentran en grupos extremos.

Por lo que la perdida del gen S podría ser un fenómeno que ocurre de forma gradual a lo largo del año. Si analizamos las secuencias que se encuentran más cercanas al cero en el eje Y se observa que se encuentran aproximadamente ordenadas por día del año. Mientras que al evaluar otras características ambientales como la duración del día se observa también un patrón ordenado. Sin embargo, la taza de cambio de la duración del día, o la rapidez con la que el día se vuelve más corto o largo, muestra una gran sincronización. Mostrando un orden de una mayor taza de cambio a una menor. Esa transición se puede observar en el genoma del virus como una disminución en el tamaño de su secuencia.

Si se observa el genoma de cada una de las secuencias aisladas, se puede percibir un pequeño desfase entre ellas. Particularmente hay regiones que muestran un mejor alineamiento entre ellas para después muestran un pequeño desfase. Este desfase se debe a la eliminación de pequeños segmentos a lo largo del genoma. La eliminación de estos pequeños fragmentos podría deberse a una adaptación al medio ambiente. Ya que esta ocurre en periodos de tiempo con una mayor duración del día.

Es importante mencionar que resulta de suma importancia la habilidad de encontrar patrones recurrentes dentro de la secuencia del SARS-Cov2. Si estos patrones se encuentran dentro de componentes estructurales que son usados como etiquetas de reconocimiento por el sistema inmune, sería posible el diseño de vacunas generalizadas para cualquier tipo de variante. El uso de péptidos que contiene la información para reconocer a varios organismos patógenos es una estrategia que ha sido utilizada de forma exitosa con anterioridad.
Por otro lado, si estos patrones recurrentes se encuentran dentro de componentes necesarios para la síntesis de la partícula viral, sería posible el tomar en cuenta esos cambios para el desarrollo de antivirales. Restringiendo la capacidad del virus para poder adaptarse a los nuevos compuestos activos.
Adaptación del genoma y quasi especie
El presente análisis se realiza con la secuencia genómica del virus. Por lo que cambios en la secuencia proteica del virus no son evaluados. Sin embargo, el análisis de la secuencia genómica evalúa cambios que no se pueden encontrar en la secuencia proteica. Estos cambios son silenciosos, ya que hay cambios en el genoma que genera secuencias que son sinónimas en proteína. Este tipo de adaptación puede deberse al tipo de codones que son usados por la célula que infecta el virus.
Todas las células expresan genes que están codificados en DNA/RNA. Particularmente la combinación de tres bases nitrogenadas genera un triplete o codón. Debido a que cada célula expresa una porción de genes distintos que la diferencian, surgen proporciones distintas de codones entre diferentes tipos de célula. Adaptarse a este cambio de proporciones facilitaría la replicación del material genético del virus. Ya que la composición del genoma viral es más similar a la composición de los genes presentes en la célula. Por lo que se requerirían un menor número de partículas virales para infectar una célula. Ademas cada codon codifica para un aminoácido especifico por lo que el virus se adaptaría también a los recursos disponibles en la célula.
Esta continua adaptación podría hacer que el genoma del SARS-Cov2 se encuentre circulando alrededor de la disponibilidad de ciertos recursos celulares. Debido a su disponibilidad se generan una serie de variantes del genoma que caen dentro de alguno de los grupos antes encontrados. Cada grupo genera una quasi especie del virus. Es decir, no hay un genoma fijo, sino que el genoma circula alrededor de un punto donde hay pequeñas variaciones entre cada copia. Posteriormente debido al proceso de transición se generan una serie de variantes hasta que nuevamente se llega a un equilibrio. Este proceso se ha repetido a lo largo de la pandemia cuando una variante desplaza a otra.
Con lo anterior se puede concluir que este tipo de representación o estructura de datos contiene la información necesaria para poder ordenas las secuencias de acuerdo a una variable ambiental. Esta variable ambiental se ha relacionado de varias formas con el COVID-19 y diversos componentes de SARS-Cov2. En el siguiente post mostraré otros análisis que muestran la relación entre la radiación solar, la duración del día y el SARS-Cov2 y el COVID-19.
Extras
S gene dropout
The spike gene target failure (SGTF) genomic signature is highly accurate for the identification of Alpha and Omicron SARS-CoV-2 variants
Codones y SARS-Cov2
Compelling Evidence Suggesting the Codon Usage of SARS-CoV-2 Adapts to Human After the Split From RaTG13
Quasi especie
El plan
El objetivo de este blog es el de conseguir fondos para mejorar y perfeccionar los métodos de análisis expuestos en “Aplications of sliding sampling to biologiocal sequences”, además de una serie de post en medium y otros análisis diversos. Puedes encontrar un índice del código y modelos propuestos en el siguiente enlace.
El desarrollo de esta metodología puede ofrecer una caracterización rápida de patógenos emergentes y otras enfermedades. Durante el desarrollo del proyecto, todos los recursos, desde código, conjuntos de datos, y ejemplos de como usar, se publicarían de modo periódico y libre.
El apoyar proyectos científicos fuera de la academia es una actividad cada vez más común, algo que se denomina ciencia ciudadana. Sin embargo, el obtener fondos para estos proyectos continúa siendo un factor limitante. El apoyar este tipo de proyectos traería a la luz nuevas ideas o ideas previamente ignoradas.
Evite las grandes aglomeraciones cerca de los puntos estacionarios de radiación solar, medio día y verano o invierno. Mejore el tiempo de exposición a la radiación solar que tiene diariamente. Y nos vemos en el siguiente.